Typ A/B ASMD
ASMD = Acid Sphingomyelinase Deficiency ist eine sehr seltene, vererbte lysosomale Speichererkrankung, der ein Mangel des Enzyms „saure Sphingomyelinase“ zugrunde liegt. Der Gendefekt ist auf dem Chromosom 11 lokalisiert (SMPD1-Gen).
Der Enzymmangel verursacht Anhäufungen von Sphingomyelin und anderen Fettstoffen in Milz, Leber, Lunge, Knochenmark und im zentralen Nervensystem (ZNS).
Man unterscheidet drei verschiedene Phänotypen (Verlaufsformen):
- Infantile neuroviszerale ASMD (M. Niemann-Pick Typ A)
- Chronisch neuroviszerale ASMD (M. Niemann-Pick Typ A/B)
- Chronisch viszerale ASMD (M. Niemann-Pick Typ B)
Historisch wurden der M. Niemann-Pick Typ A/B und M. Niemann-Pick Typ C als eine Erkrankung aufgefasst. M. Niemann-Pick Typ A/B und Typ C unterscheiden sich jedoch genetisch, biochemisch und klinisch voneinander.
Zur Unterscheidung des M. Niemann-Pick Typ A/B vom M. Niemann-Pick Typ C wird international die Verwendung einer überarbeiteten Nomenklatur empfohlen.
ASMD steht für den englischen Begriff „ACID SPHINGOMYELINASE DEFICIENCY“, übersetzt bedeutet dies „Saure Sphingomyelinase-Mangel“.
| Historische Einordnung | Empfohlene neue Nomenklatur |
| M. Niemann-Pick Typ A | Infantile neuroviszerale ASMD |
| M. Niemann-Pick Typ A/B | Chronisch neuroviszerale ASMD |
| M. Niemann-Pick Typ B | Chronisch viszerale ASMD |
ASMD – die 3 Subtypen
-
Typ A (ASMD)
-
Typ B (ASMD)
-
Typ A/B (ASMD)
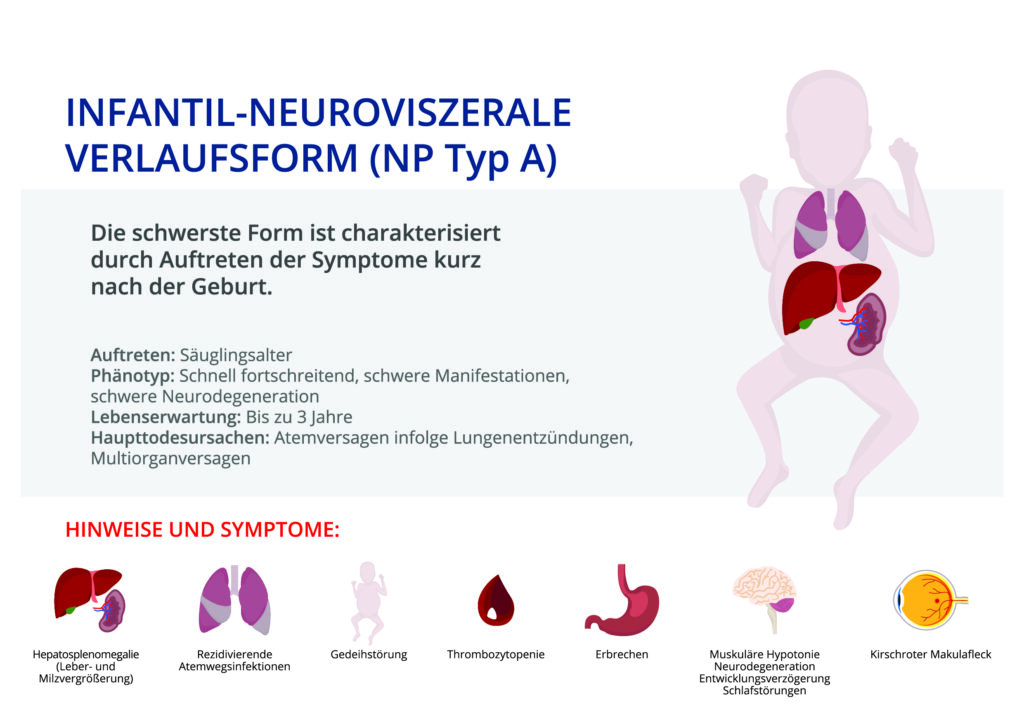
Symptome wie Schwierigkeiten bei der Fütterung, Wachstumsstörungen und ein deutlich vergrößerter Bauch (aufgrund Organvergrößerungen — ) können auftreten. Die Lebenserwartung liegt in etwa bei 3 Jahren.
Die infantile neuroviszerale ASMD (M. Niemann-Pick Typ A) ist eine sehr schwere, neurodegenerative Form der Erkrankung, die bereits im Säuglingsalter beginnt. Eine ausgeprägte Leber – und Milzvergrößerung ist meist das erste festgestellte Symptom. Kinder mit der infantilen neuroviszeralen ASMD können auffällige Gesichtsmerkmale aufweisen, wie vergröberte Gesichtszüge und Gesichtsschwellungen. Ca. 50 % der betroffenen Kinder haben einen kirschroten Makulafleck („Cherry red spot“), dabei handelt es sich um Lipidablagerungen in der Netzhaut. Obwohl eine muskuläre Hypotonie (verminderte Muskelspannung, Muskelschwäche) bereits in den ersten Lebensmonaten auftreten kann, entwickeln sich die betroffenen Kinder in der Regel bis zum 6. Lebensmonat normal. Zwischen dem 6. und 15. Lebensmonat kommt es zu einem Entwicklungsstillstand, gefolgt von einer schnell fortschreitenden psychomotorischen Verschlechterung. Die meisten Kinder entwickeln nie die Fähigkeit frei zu sitzen, zu krabbeln oder zu laufen. Ein weiteres Krankheitszeichen ist eine Gedeihstörung. Schwierigkeiten bei der Fütterung aufgrund einer Saugschwäche und Magenkompression verschlimmern im Verlauf die Gedeihstörung. Die Kinder entwickeln häufige Atemwegsinfekte aufgrund von Aspirationen (Eindringen von flüssiger oder fester Nahrung in die Atemwege, „Verschlucken“). Die schnelle und fortschreitende Neurodegeneration (Untergang von Nervenzellen des zentralen Nervensystems) führt zu einem frühen Tod der Kinder. Die häufigste Todesursache ist Atemversagen infolge von Lungenentzündungen.
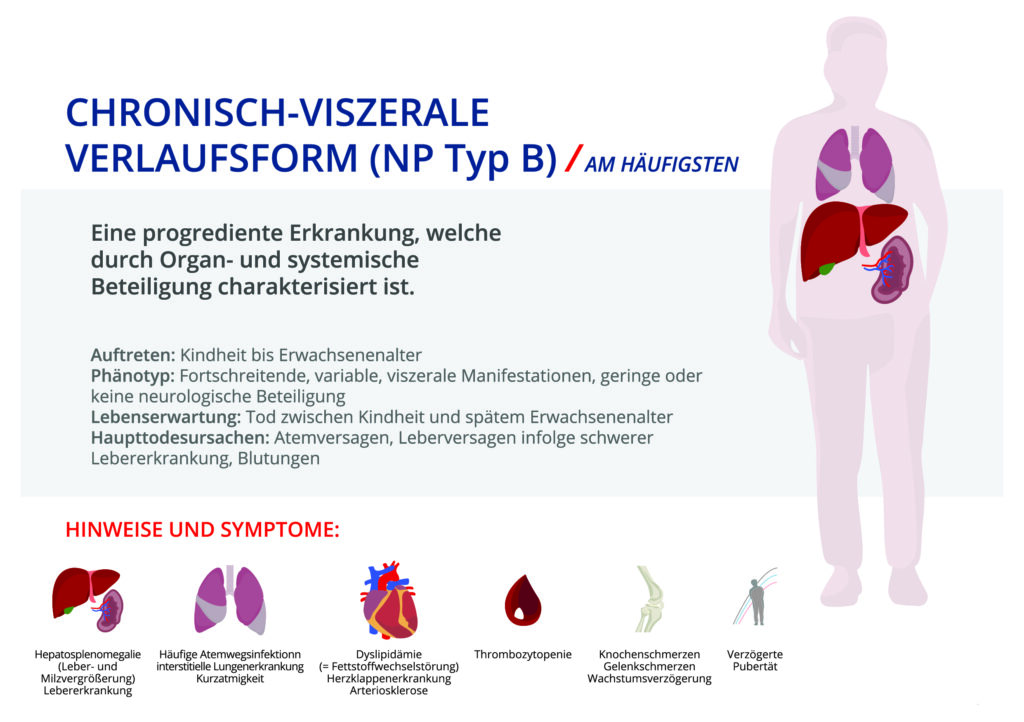
Tyb B basiert ebenfalls auf demselben Enzymmangel, jedoch ist hier eine Restaktivität von ca. 3-10 % vorhanden. Dadurch ist eine schwere Beeinträchtigung des zentralen Nervensystems auszuschließen.
Bei der chronisch viszeralen ASMD (M. Niemann-Pick Typ B) können Schwere und Ausmaß der Symptome stark variieren. Erste Symptome sind meist eine Leber- und Milzvergrößerung. Weitere Manifestationen können eine interstitielle Lungenerkrankung, Thrombozytopenie und erhöhte Blutfettwerte sein. Bei dieser Form besteht keine bis geringe Beteiligung des zentralen Nervensystem. Der Krankheitsverlauf ist variabel.
Zu den Symptomen gehören Wachstumsverzögerung, progressive Organvergrößerung von Milz und und Leber, hohe Anfälligkeit für Atemwegsinfektionen, Blutungsneigung (Thrombozytopenie), Knochenschmerzen und Belastung des Herzens. Die Patienten können ein hohes Erwachsenenalter erreichen.
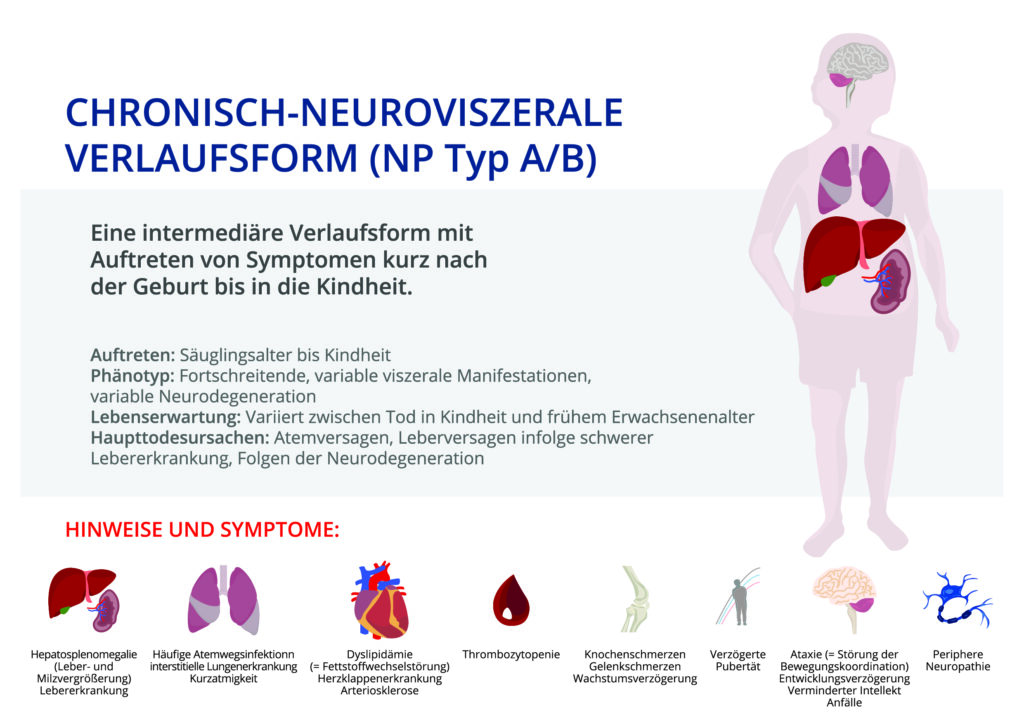
Die chronisch neuroviszerale ASMD (M. Niemann-Pick Typ A/B) ist eine Mischform aus Typ A und B. Zusätzlich zu den Symptomen der chronisch viszeralen ASMD treten neurologische Symptome im Verlauf auf, die langsam fortschreiten. Zu den neurologischen Krankheitszeichen gehören eine geistige und motorische Entwicklungsverzögerung /-störung, eine Ataxie (Störung der Bewegungskoordination) und eine Störung des peripheren Nervensystems.
ASMD-Aufklärungsvideo
In diesem Video werden die 3 Typen von ASMD und ihre Verlaufsformen veranschaulicht. Zudem gibt es Informationen über die Entstehung der Erkrankung und einen Einblick in den Zellprozess verursacht durch den Enzymmangel. Weltweit kommt ASMD bei 1:250.000 Menschen vor.
Unterstützen Sie uns und teilen sie das Video für mehr Aufklärung für diese sehr seltene Erkrankung.
(bitte anklicken)

LYSOSOLUTIONS (Sanofi)
Ausführliche Informationen über die Erkrankung, Symptome und Diagnostik sowie Illustrationen und Videos klären über ASMD auf.
Unter Punkt 6 sind auch Erfahrungsberichte von betroffenen Patient:innen zu sehen.